Fenilcetonuria: Un trastorno genético en el gen PAH
Está asociada con mutaciones en el gen PAH, el cual proporciona instrucciones para producir una enzima llamada fenilalanina hidroxilasa.
La fenilcetonuria (PKU, por sus siglas en inglés) es un trastorno genético raro que afecta la capacidad del cuerpo para descomponer el aminoácido fenilalanina, que se encuentra en proteínas y en ciertos alimentos. Si no se trata, la acumulación de fenilalanina en el cuerpo puede causar daño cerebral y otros problemas de salud. Conocer las opciones de tratamiento es crucial para el manejo de todas las complicaciones.
Enfermedades genéticas: Los diferentes tipos y su impacto
Causas de la fenilcetonuria
Es causada por mutaciones en el gen PAH, que proporciona instrucciones para producir la enzima fenilalanina hidroxilasa. Estas mutaciones pueden variar en gravedad, lo que afecta la cantidad y la actividad de la enzima producida. Sin suficiente cantidad de enzimas, el cuerpo no puede descomponer adecuadamente la fenilalanina, lo que lleva a su acumulación en la sangre y el cerebro.
Te recomendamos este videoLa PKU es hereditaria y se transmite de padres a hijos a través de un patrón autosómico recesivo. Esto significa que ambos padres deben portar el gen defectuoso para que su descendencia desarrolle la enfermedad.
Síndrome de Marfan: Una condición genética particular
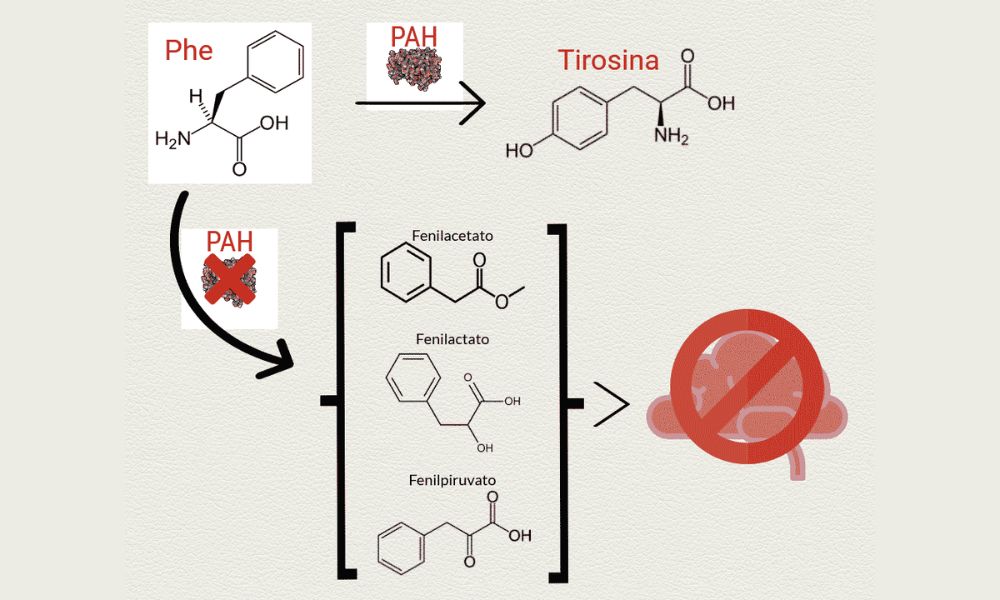
Síntomas de la fenilcetonuria
Los síntomas pueden variar dependiendo del grado de la enfermedad, pero generalmente incluyen:
- Falta de desarrollo normal.
- Problemas de alimentación.
- Irritabilidad.
- Piel, cabello y ojos de color claro.
- Olor inusual en la respiración, piel o orina, a menudo descrito como “olor a ratón”.
- Retraso en el desarrollo.
- Problemas de comportamiento.
- Problemas de aprendizaje.
- Convulsiones.
- Piel pálida.
- Hiperactividad.
- Problemas en el corazón.
- Problemas en la piel.
- Problemas de movimiento.
Es importante tener en cuenta que algunos bebés pueden no presentar síntomas evidentes al nacer, lo que subraya la importancia del cribado neonatal para detectar la afección.
¿Cómo se diagnostica la PKU?
Se diagnostica principalmente a través de pruebas de detección neonatal y pruebas confirmatorias.
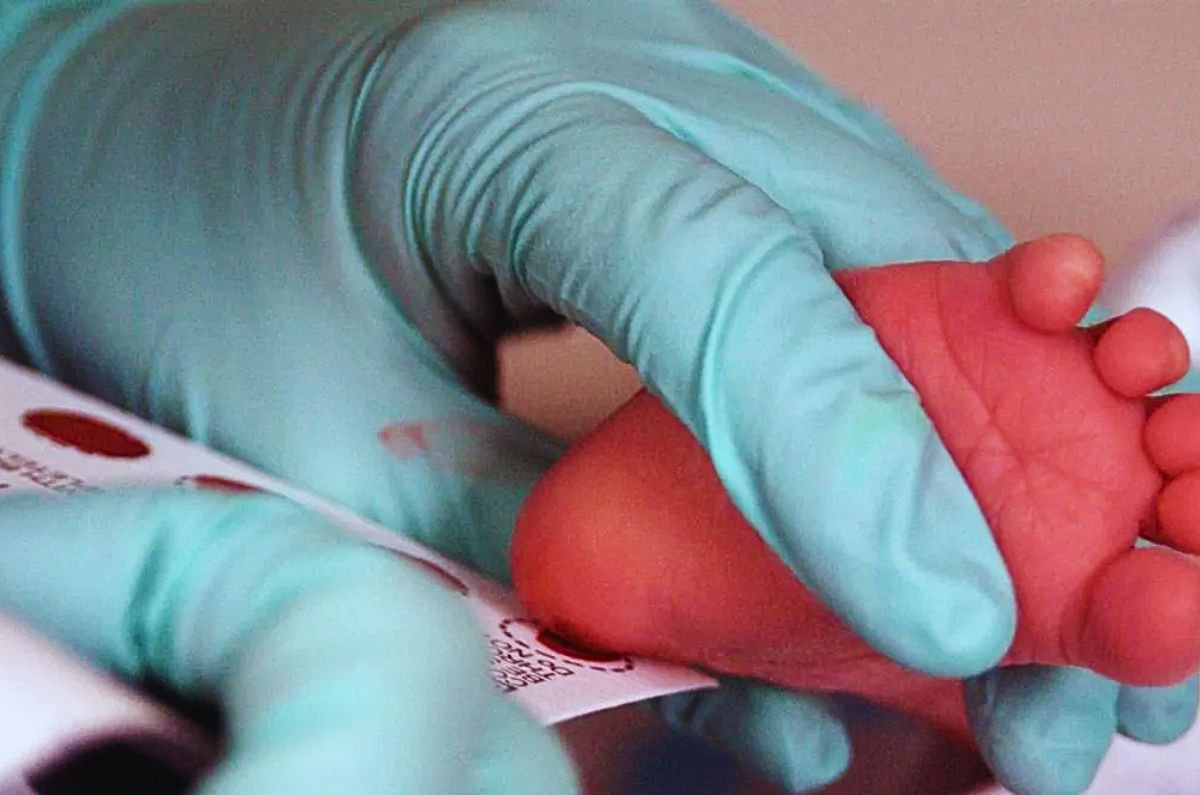
- Cribado neonatal: se toma una pequeña muestra de sangre del talón del recién nacido, generalmente entre las 48 horas después del nacimiento. Posteriormente, se analiza para medir los niveles de fenilalanina. Niveles elevados pueden indicar la presencia de PKU.
- Pruebas confirmatorias: si una prueba de detección inicial muestra niveles elevados de fenilalanina, se realiza un diagnóstico adicional para confirmar.
- Prueba de orina: se puede realizar una prueba de orina para medir los niveles de metabolitos de la fenilalanina.
- Prueba genética: para detectar mutaciones en el gen responsable de la PKU.
Síndrome de Turner: Un trastorno genético en mujeres
Tratamiento para la fenilcetonuria: Dieta
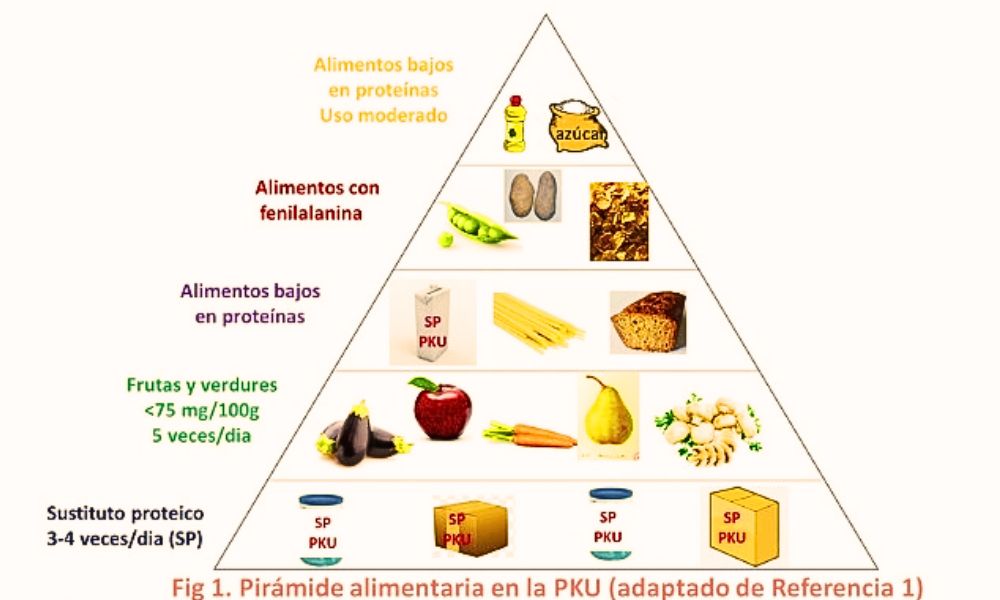
El tratamiento primario para la PKU implica seguir una dieta baja en fenilalanina. Esto implica evitar alimentos ricos en proteínas que contienen altos niveles, como carne, pescado, huevos, productos lácteos, nueces y legumbres. En su lugar, se deben consumir fórmulas especiales bajas en fenilalanina y alimentos diseñados específicamente para personas con la enfermedad, los cuales pueden incluir harinas especiales, mezclas para hornear, pastas, entre otros.
Consecuencias de la fenilcetonuria no tratada
Sin un tratamiento o manejo adecuado, la PKU puede tener varias consecuencias negativas para la salud:
- Retraso mental: puede haber daño cerebral, lo que lleva al retraso si no se trata desde una edad temprana.
- Problemas de comportamiento: los niveles elevados de fenilalanina suelen provocar problemas como hiperactividad, irritabilidad y problemas de atención.
- Trastornos en el desarrollo: los niños no tratados con PKU pueden experimentar retraso en el crecimiento y problemas en su desarrollo.
- Problemas en la piel: la acumulación de fenilalanina puede causar erupciones cutáneas y decoloración de la piel.
- Daño en el corazón y los riñones: los niveles altos de fenilalanina pueden dañar el corazón y los riñones con el tiempo.
Tratar la PKU desde una edad temprana con una dieta baja en fenilalanina es crucial para prevenir estas consecuencias y permitir un desarrollo saludable.
Neurofibromatosis: Una guía completa para entender este trastorno genético
La fenilcetonuria es un trastorno metabólico hereditario que afecta la forma en que el cuerpo descompone y utiliza la fenilalanina. Con una detección temprana y un tratamiento adecuado, las personas con PKU pueden llevar una vida saludable y minimizar el riesgo de complicaciones a largo plazo. Es fundamental contar con el apoyo de profesionales médicos y nutricionistas especializados para garantizar un manejo efectivo de la enfermedad y optimizar la calidad de vida de quienes viven con esta afección.









{kind=link}