Síndrome de Apert: Los desafíos de esta rara condición genética
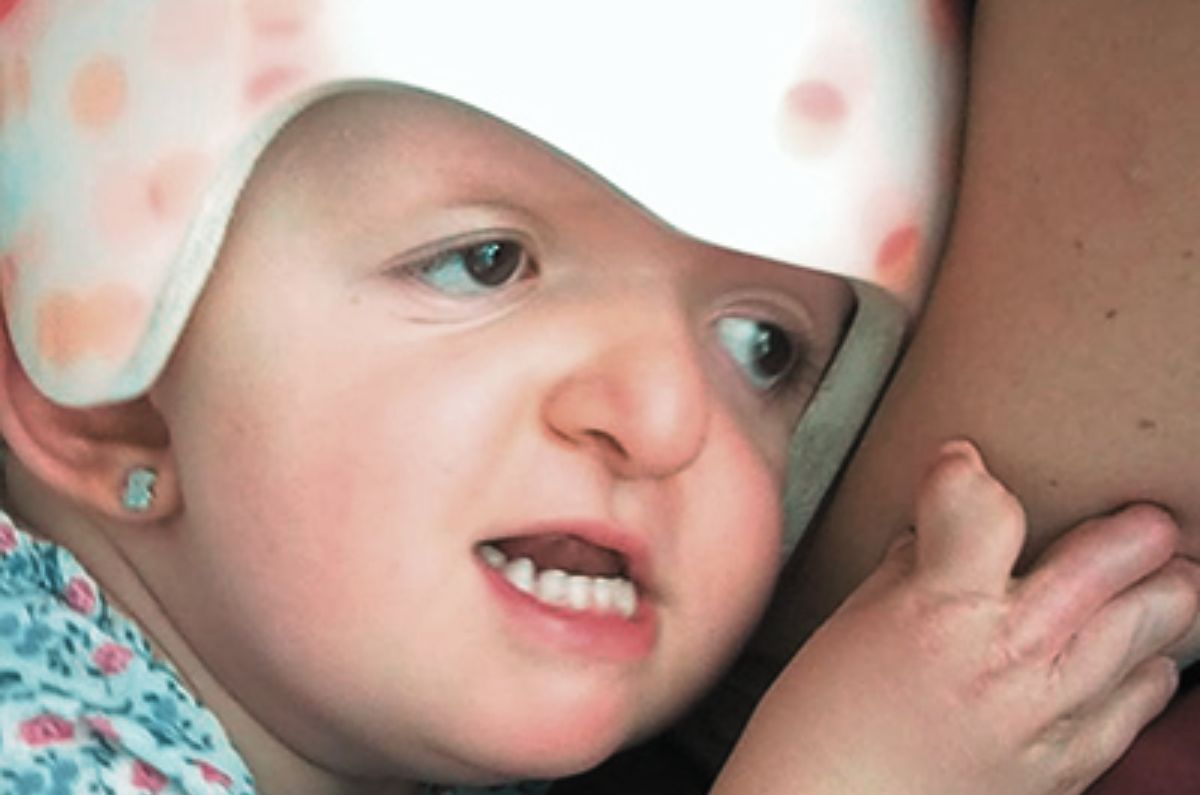
Es un tipo de craneosinostosis sindrómica, descrita a inicios de 1900 por el pediatra francés Eugène Apert.
El síndrome de Apert es una rara condición genética que afecta a aproximadamente 1 de cada 65,000 a 88,000 recién nacidos en todo el mundo. Esta condición, también conocida como acrocefalosindactilia, se caracteriza por el cierre prematuro de las suturas del cráneo y una fusión anormal en las manos y los pies, comúnmente en los dedos. Es importante conocer a profundidad esta enfermedad genética para saber cómo afrontar los diferentes desafíos que conlleva.
Síndrome de Lynch: El origen genético de ciertos tipos de cáncer
Causas del síndrome de Apert
La acrocefalosindactilia es causada por una mutación genética espontánea que ocurre durante el desarrollo embrionario, la cual afecta al gen FGFR2: el responsable de regular el crecimiento y desarrollo de los huesos y tejidos del cuerpo. Esta mutación provoca una activación anormal de las vías de señalización celular, lo que afecta el desarrollo craneofacial y esquelético.
Este video te puede interesar
En la mayoría de los casos, el síndrome no es hereditario, sino que es el resultado de una mutación nueva en el gen FGFR2. Sin embargo, en ciertas situaciones, la mutación puede ser heredada de uno de los padres que presenta el mismo gen alterado.
Síntomas de la acrocefalosindactilia
Los síntomas pueden variar en severidad de una persona a otra, pero generalmente incluyen:
- Síntomas craneofaciales:
- Craneosinostosis: cierre prematuro de las suturas del cráneo.
- Forma anormal de la cabeza.
- Frente prominente y ojos ampliamente separados.
- Nariz chata y puente nasal bajo.
- Mandíbula pequeña y prognatismo.
- Síntomas en manos y pies:
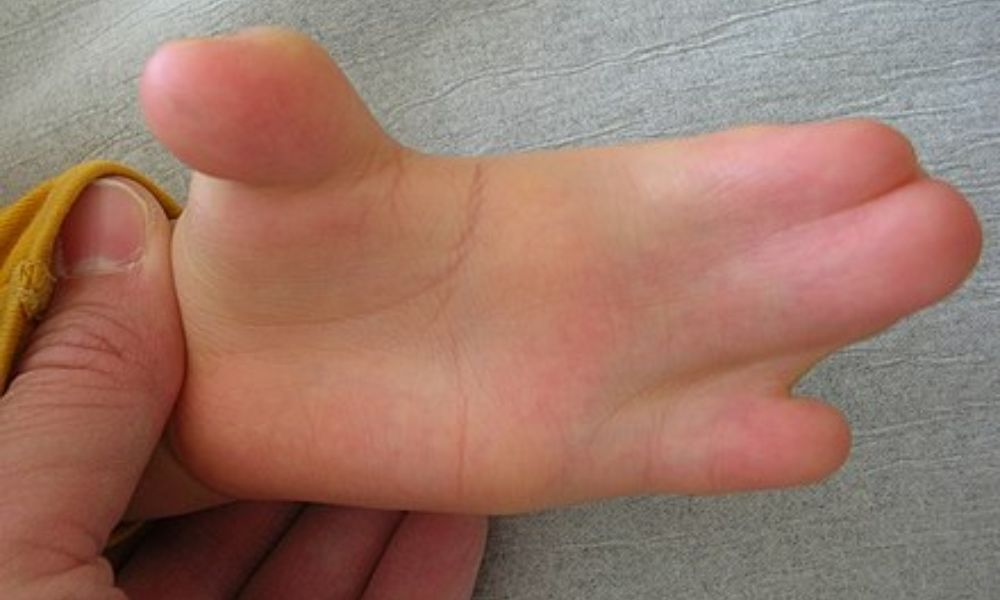
- Sindactilia: fusión de los dedos de las manos y los pies.
- Dedos anormalmente cortos y en forma de pulgar.
- Otros síntomas:
- Problemas dentales.
- Problemas de audición.
- Retraso en el desarrollo cognitivo.
Consumir fibra puede disminuir el riesgo de deterioro cognitivo: Estudio

Diagnóstico del síndrome de Apert
El diagnóstico generalmente se realiza a través de una combinación de evaluación clínica, pruebas de imágenes y análisis genéticos.
- Evaluación clínica
- Examen físico: un médico puede identificar características físicas distintivas asociadas con el síndrome, como la forma anormal del cráneo y la cara, y anomalías en las manos y los pies.
- Historia médica: se obtiene información detallada sobre el desarrollo y los antecedentes familiares del paciente.
- Pruebas genéticas
- Análisis molecular: se puede realizar una prueba genética para buscar la mutación genética específica asociada con el síndrome. Esto generalmente implica extraer una muestra de sangre u otra muestra de tejido para el análisis.
- Evaluación adicional:
- Radiografías: se realizan en el cráneo, las manos y los pies para evaluar cualquier anomalía estructural.
- Audiometrías: son pruebas importantes debido a que algunos individuos con este síndrome pueden experimentar pérdidas auditivas.
- Evaluación de otros órganos: en algunos casos, se pueden realizar pruebas adicionales para evaluar si hay anomalías en otros órganos o sistemas del cuerpo.
Es importante buscar atención médica de profesionales especializados en genética y trastornos craneofaciales para obtener un diagnóstico preciso.
Fenilcetonuria: Un trastorno genético en el gen PAH
Tratamiento para el síndrome de Apert
El tratamiento es multidisciplinario y se enfoca en abordar los diferentes aspectos de la condición. La cirugía es una parte importante y se puede realizar para corregir las fusiones anormales de los huesos en las manos y los pies, así como para remodelar el cráneo y mejorar la apariencia facial. Además, las personas con síndrome pueden requerir terapia física y ocupacional para mejorar la función y movilidad de las extremidades, así como terapias específicas para abordar los problemas de audición y el desarrollo del habla.
Esperanza de vida en personas con síndrome de Apert
La esperanza de vida ha mejorado significativamente en las últimas décadas gracias a los avances en la atención médica y quirúrgica. Sin embargo, cada caso es único y la salud puede verse afectada por la gravedad de los síntomas y cualquier complicación médica adicional que pueda estar presente. Es importante destacar que, aunque no es curable, con un tratamiento y manejo adecuados muchos pacientes suelen llevar vidas saludables y productivas.
Síndrome de Rett: Una rara condición genética
Desafíos y dificultades del síndrome de Apert
El síndrome presenta una serie de desafíos y dificultades para las personas que lo padecen y sus familias. Los problemas de salud asociados, como la dificultad para respirar, problemas dentales y auditivos, y retraso en el desarrollo, pueden requerir atención médica y terapéutica a largo plazo. Además, los rasgos faciales distintivos y las fusiones anormales en las extremidades suelen afectar la autoestima y la calidad de vida de los pacientes. Es importante proporcionar un apoyo integral y multidisciplinario para abordar cada complicación y promover el bienestar de quienes viven con esta condición.

A pesar de ser una condición genética rara, el síndrome de Apert puede ser manejado con un enfoque multidisciplinario que incluye cirugía, terapias físicas y ocupacionales, así como evaluaciones médicas especializadas. Con el tratamiento adecuado, muchas personas afectadas pueden llevar vidas saludables y productivas. Es importante consultar a un equipo médico experimentado en el manejo de la acrocefalosindactilia para desarrollar un plan de manejo individualizado y recibir un seguimiento particular.